Correlative FIB-SEM and TEM investigation of the Mg electrode cycling mechanism
- Abstract number
- 317
- Presentation Form
- Contributed Talk
- Corresponding Email
- [email protected]
- Session
- EMAG - Energy Materials
- Authors
- Mr Andrzej Sankowski (1), Mr Konstantinos Dimogiannis (1), Dr Conrad Holc (1), Dr Christopher Parmenter (1), Dr Graham Newton (1), Dr Darren Walsh (1), Dr James O'Shea (1), Prof Andrei Khlobystov (1), Dr Lee Johnson (1)
- Affiliations
-
1. University of Nottingham
- Keywords
Focused-ion beam, magnesium-ion battery magnesium battery, Mg(TFSI)2, glyme ethers, solid electrolyte interphase
- Abstract text
The magnesium battery is unique in its ability to exceed the volumetric energy density of lithium-based batteries. However, during cycling of magnesium electrodes in practical electrolytes, large coulombic inefficiencies are observed and significant overpotentials must be applied to drive electron transfer. Understanding the origin of these problems is one of the major challenges in this field and requires a detailed knowledge of the composition of Mg electrodes, which is unavailable. In this work, the detailed chemical and structural changes that occur during Mg cycling in Mg(TFSI)2-glyme electrolyte solutions, the base solution used in many studies, are described for the first time. Using focused ion beam-scanning electron microscopy, we determine the three-dimensional chemical composition of this Mg electrode upon electrochemical cycling. We show that the Mg electrode consists of three discrete layers associated with interphase formation, degradation and active Mg. This structure undergoes expansion and contraction during cycling due to incorporation of Mg into the middle layer, resulting in structural deformation and degradation. The electrochemical performance is elucidated based on this structural model and we demonstrate that the initial high overpotential during cycling is due to the nucleation barrier for Mg.
The lithium-ion battery (LIB) has been at the forefront of innovations in energy storage since its commercialisation over 30 years ago. As we reach the practical specific capacity limit of current state-of-the-art materials, novel and innovative battery technologies are required to satiate the requirements of energy intensive applications such as electric vehicles.1 The use of a lithium metal negative electrode has been proposed to increase the energy density, however the high reactivity causes poor cycling performance due to electrolyte degradation, dendrite formation and safety issues.2 The magnesium electrode offers a solution that has double the theoretical volumetric capacity of the lithium electrode while simultaneously mitigating dendritic growth, reducing raw material costs and greatly increasing sustainability.3,4 Previous work has shown that traditional LIB electrolytes are not compatible with magnesium metal, and the plating and stripping mechanism that underpins how magnesium electrodes cycle is poorly understood.5
Here we explored the mechanism of magnesium plating and stripping in glyme-based electrolytes with a range of additional additives. We show that in the pure electrolyte solution, interphase formation is critical to stable cycling but that this is accompanied by significant degradation and accumulation of inactive Mg.6 We establish the three-dimensional chemical composition of Mg deposits in the different electrolyte formulations using a combination of focused ion beam-scanning electron microscopy (FIB-SEM), energy dispersive x-ray spectroscopy (EDX) and transmission electron microscopy (TEM). We use these findings to develop a mechanistic understanding that explains the electrochemical cycling performance of magnesium metal negative electrodes. In situ electrochemical quartz crystal microbalance (EQCM) measurements are used to evaluate the plating and stripping efficiency of the electrodes and to identify non-electrochemical degradation reactions between Mg and the electrolyte solutions.
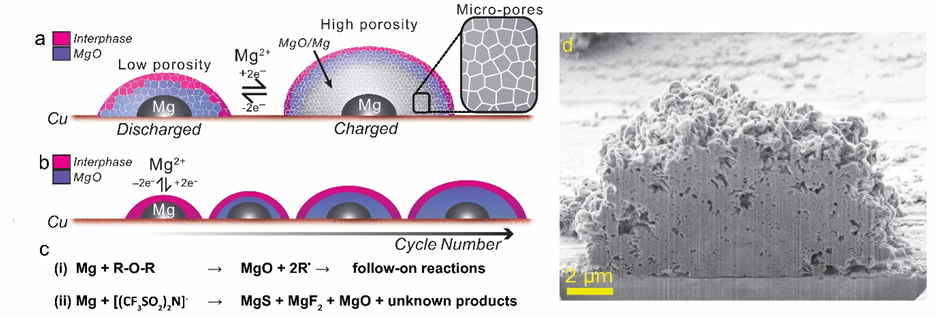
Figure 1. Schematic of the (a) the key chemical and structural transformations occurring during cycling of the Mg electrode and (b) the growth of the MgO outer core due to accumulation of dead Mg, as MgO over successive cycles. Expanded region shows the micro and macro pores that permeate the Mg deposit. (c) Degradation reactions occurring between the electrolyte solution and Mg that lead to formation of the interphase and MgO outer core. (d) A cross section of a discharged deposit showing the porosity along with the inner (lighter) and outer (darker) core layers.
By combining electrochemical methods with imaging and analysis at the nanoscale by FIB-SEM, TEM, e-beam diffraction and surface spectroscopy, the chemical and structural changes that occur during cycling of the Mg electrode in the base electrode Mg(TFSI)2-glyme have been described in detail for the first time. During charging, Mg forms dome-shaped particles with a nanocrystalline surface and porous volume. A cross-sectional analysis revealed a complex, onion-like internal structure, consisting of a Mg metal inner core, wrapped in an insulating Mg/MgO outer core, and an inorganic Mg salts (MgS/MgF2) interphase layer. Mg metal grows from a discrete metallic inner core and is incorporated within a MgO outer core resulting in a ~400% volume expansion and contraction during cycling. The data suggests that even relatively stable ethers undergo degradation resulting in accumulation of dead Mg as MgO and an ever increasing electrode volume, the mass of which is measured via EQCM measurement. These data provide a critical understanding of the problems of the Mg electrode and a foundation to which the impact of new additives and salts can be compared.
- References
1 R. Shah, V. Mittal, E. Matsil and A. Rosenkranz, Adv. Mech. Eng., 2021, 13, 1–9.
2 Z. Hu, J. Li, X. Zhang and Y. Zhu, Front. Chem., 2020, 8, 409.
3 M. Fichtner, Magnesium Batteries: Research and Applications, Royal Society of Chemistry, 2019, vol. 2020.
4 R. Attias, M. Salama, B. Hirsch, Y. Goffer and D. Aurbach, Joule, 2019, 3, 27–52.
5 S. B. Son, T. Gao, S. P. Harvey, K. X. Steirer, A. Stokes, A. Norman, C. Wang, A. Cresce, K. Xu and C. Ban, Nat. Chem., 2018, 10, 532–539.
6 C. Holc, K. Dimogiannis, E. Hopkinson and L. R. Johnson, ACS Appl. Mater. Interfaces, 2021, 13, 29708–29713